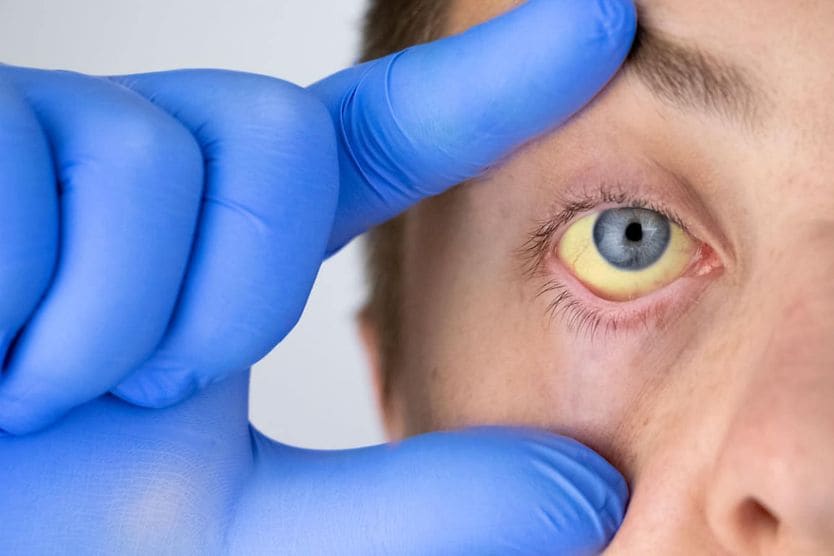
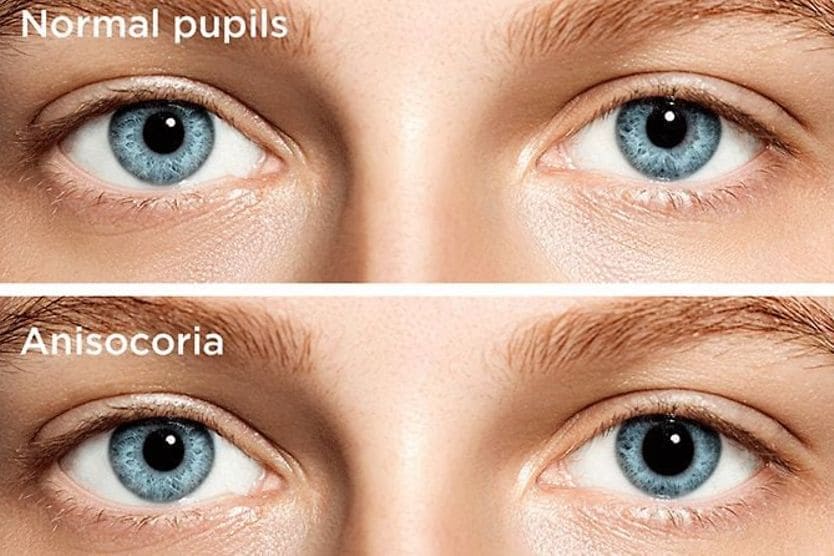
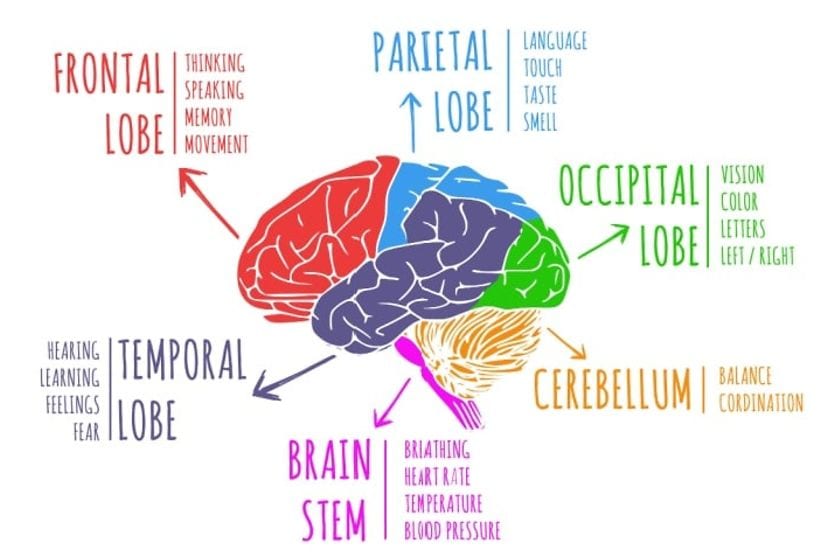
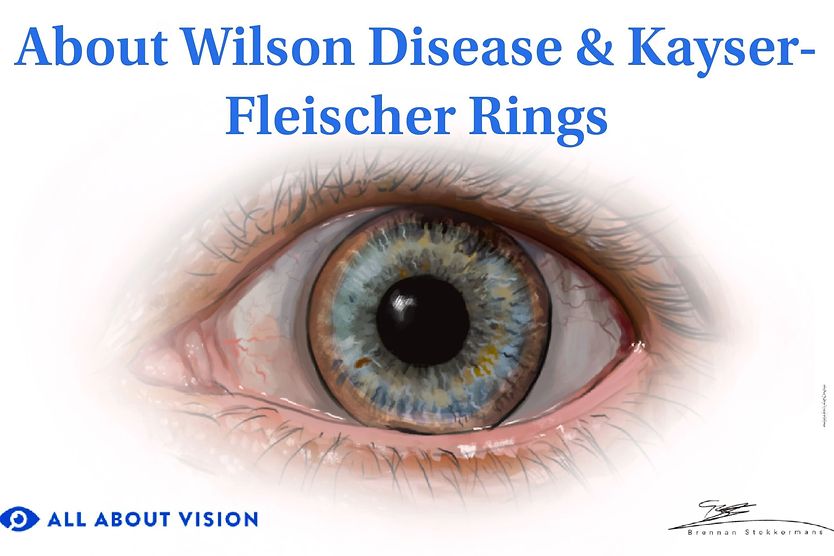
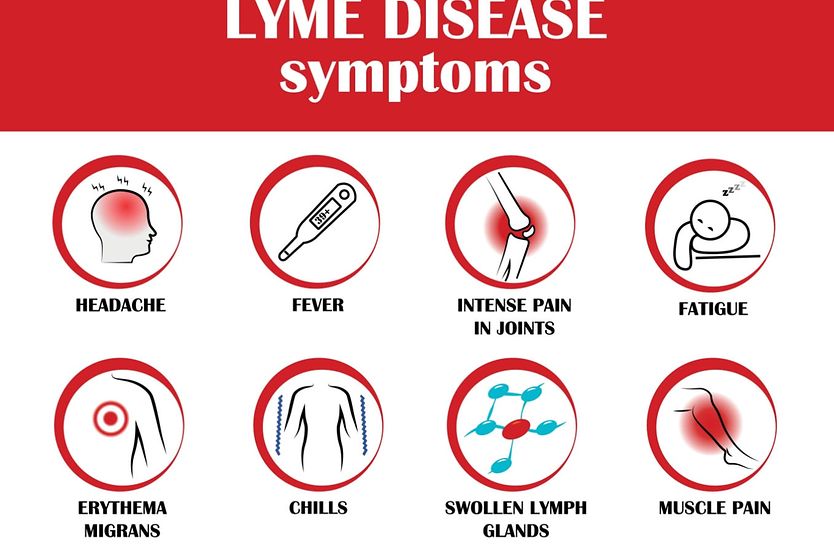
Related Medical Conditions
Often, issues with our eyes can stem from something else that may seem totally unrelated. Learn about some of the underlying conditions can have an effect our eyes
Related Articles












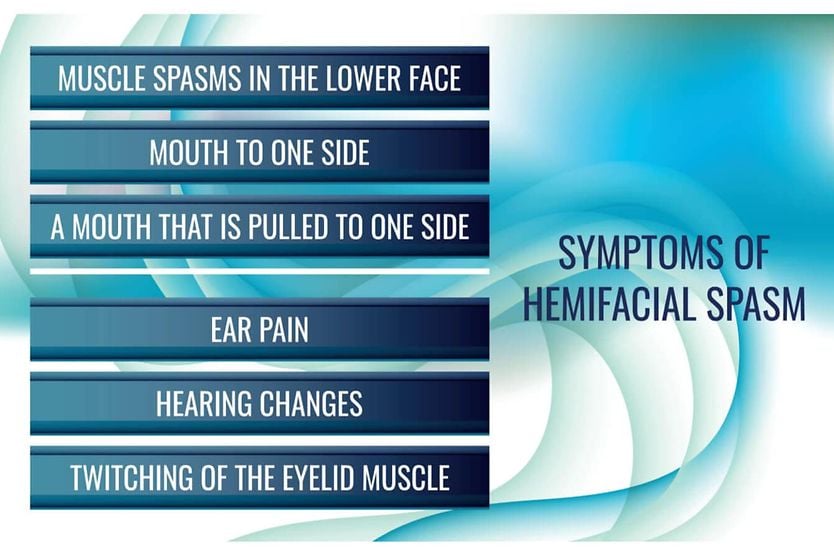



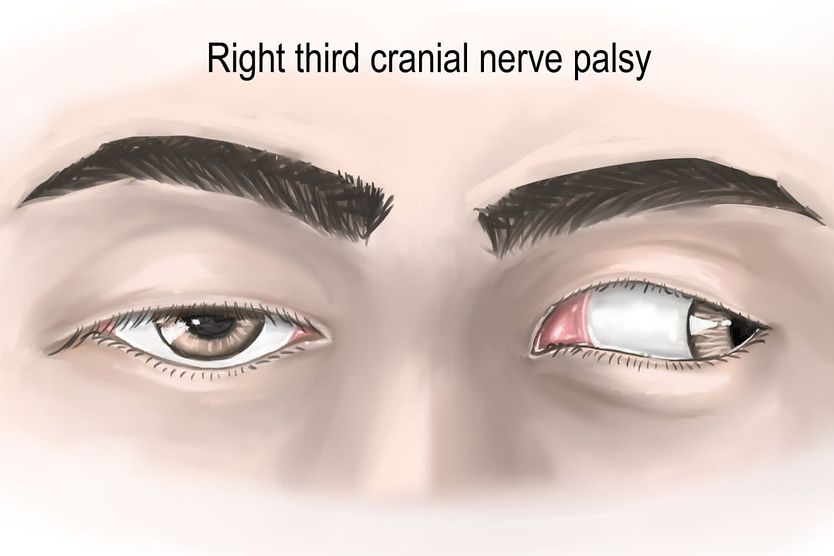









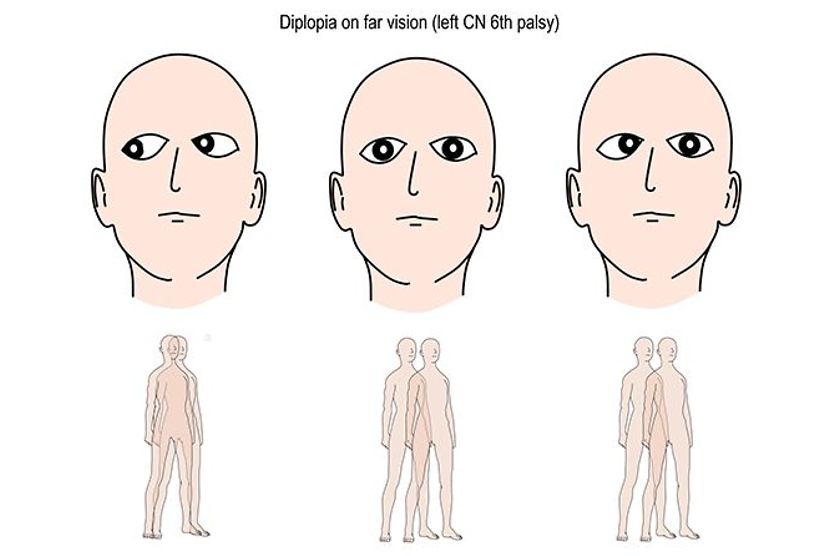
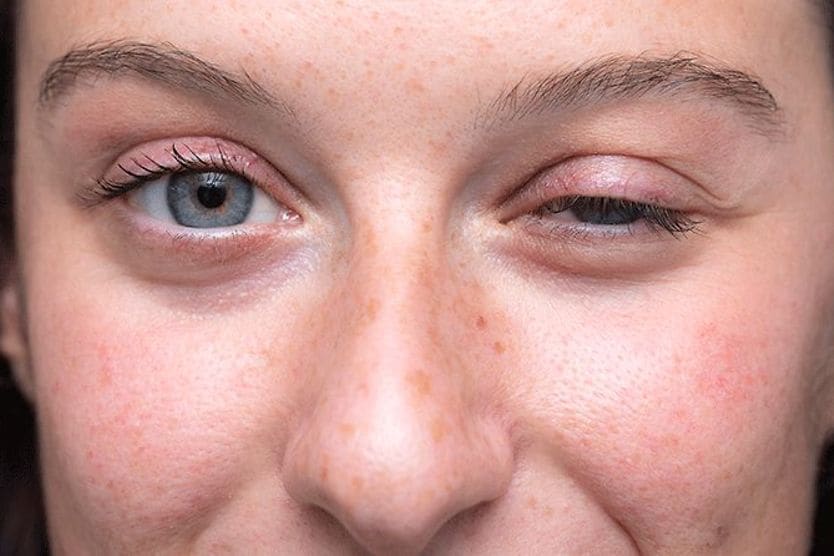
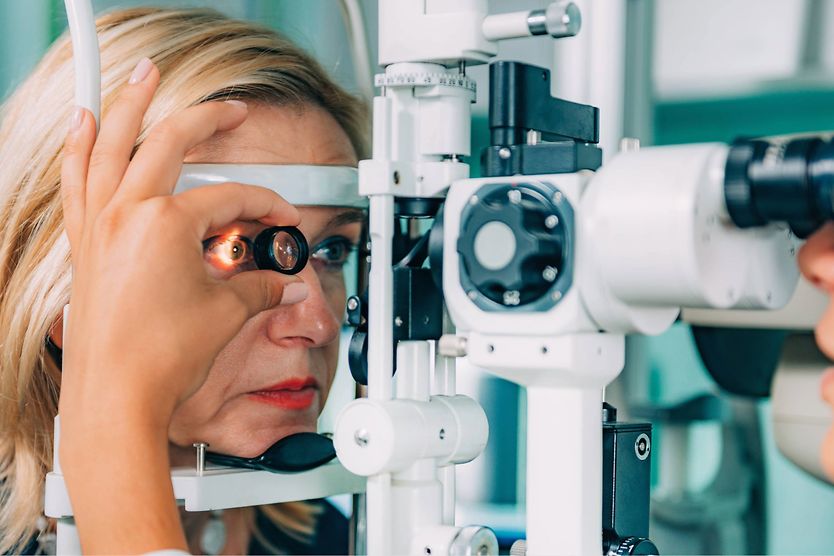
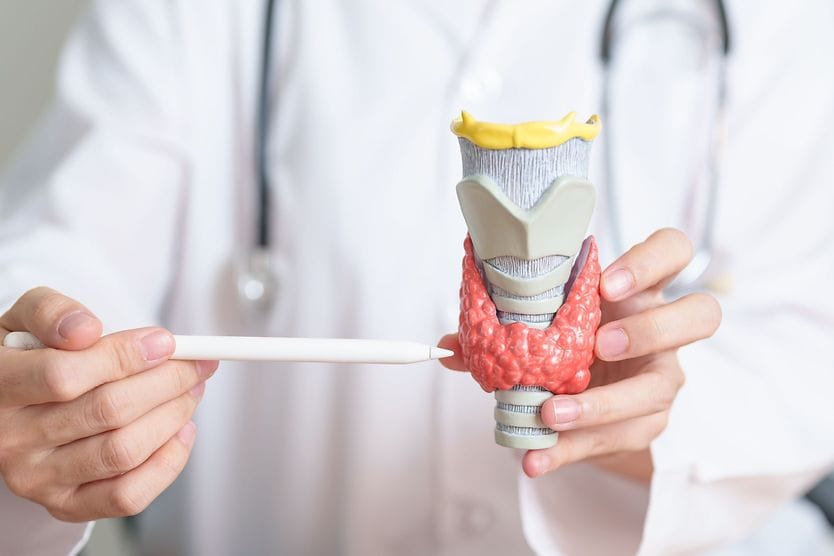

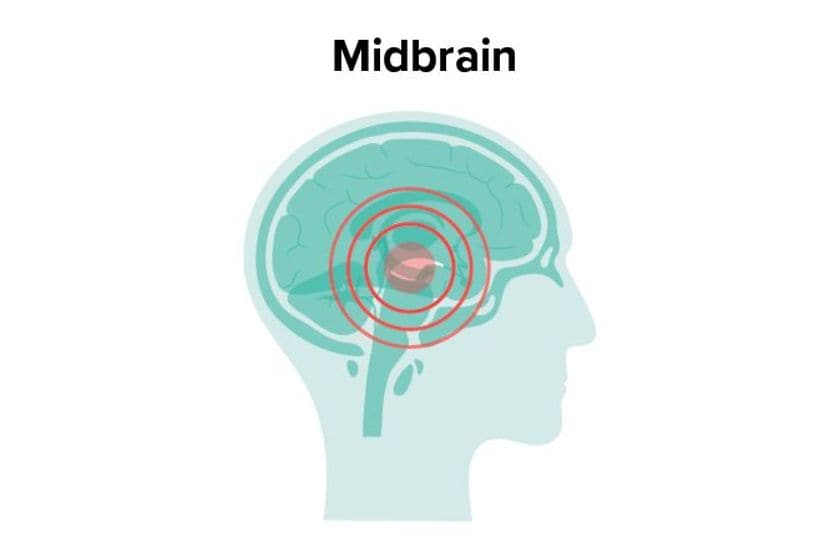
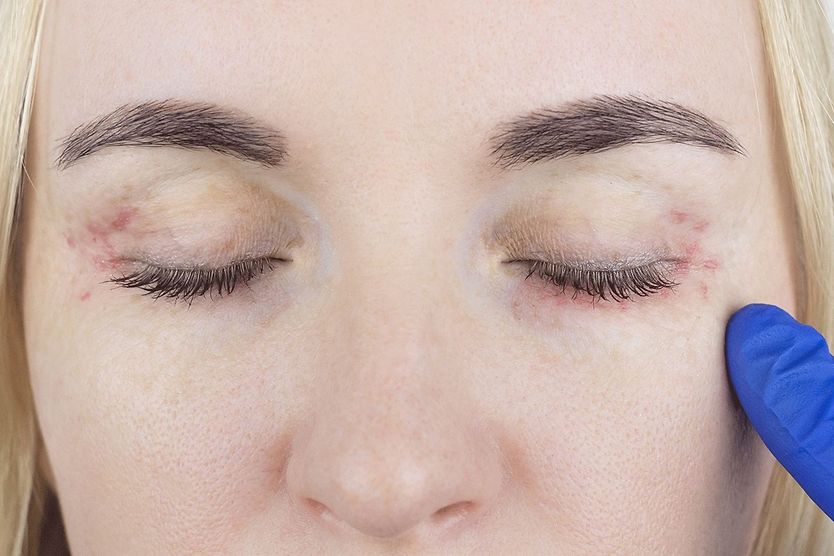





















All About Vision and AllAboutVision.com are registered trademarks of Essilor Laboratories of America, Inc © 2000-2026 Essilor Laboratories of America, Inc. The content on this site is for informational purposes only. All About Vision does not provide medical advice, diagnosis or treatment. Contact an eye doctor if you need medical attention.