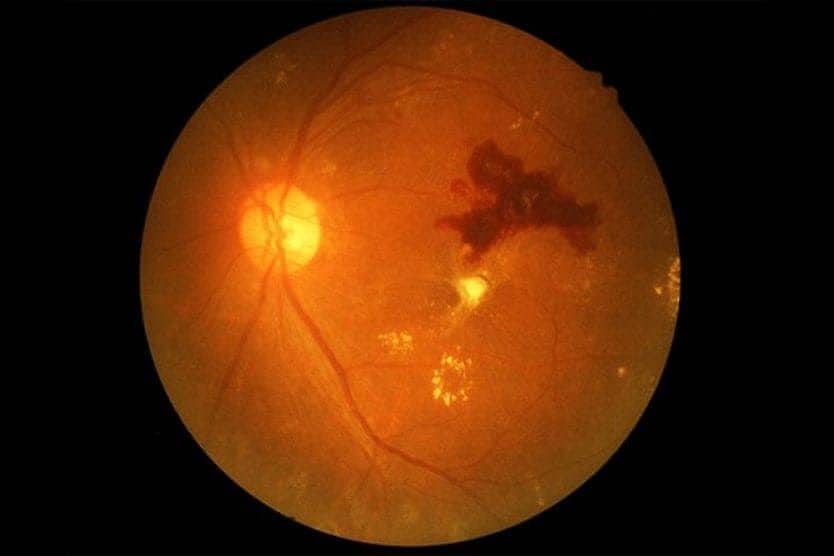
Retinitis pigmentosa

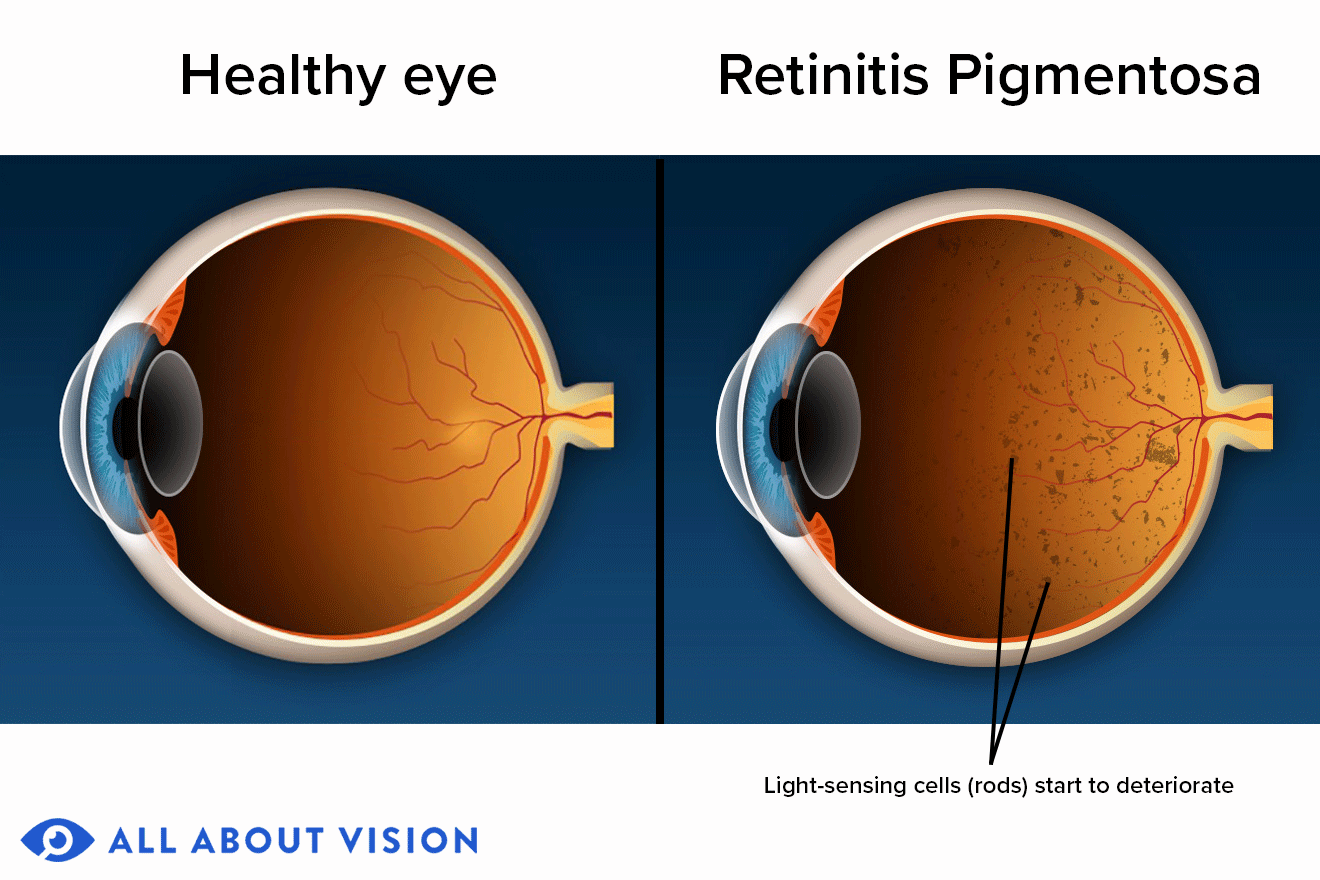
Retinitis pigmentosa (RP) is a rare, inherited disease in which the light-sensitive retina of the eye slowly and progressively degenerates. Eventually, blindness can result.
When retinitis pigmentosa is suspected, visual field testing likely will be conducted during or after your comprehensive eye exams to determine the extent of peripheral vision loss. Other specialized eye tests may be needed to determine whether you have lost night vision or color vision.
Symptoms of retinitis pigmentosa
The first signs of retinitis pigmentosa usually occur in early childhood, when both eyes typically are affected. Night vision can be poor, and the field of vision may begin to narrow.
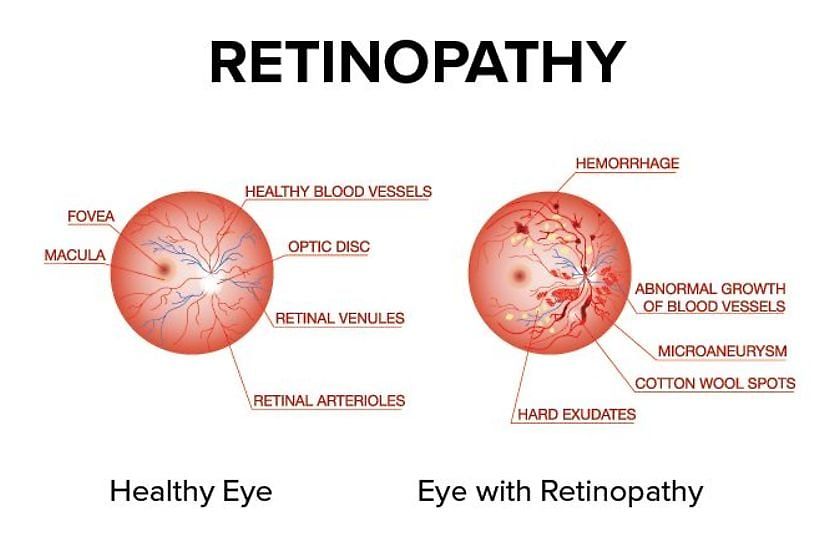
Retinal pigmentation is a sign of deterioration of light-sensitive cells: it can be difficult to see clearly in low light.
When RP first starts to appear, the light-sensing cells that are responsible for vision in dim light (rods) gradually deteriorate, and seeing at night often becomes more difficult.
During later stages of retinitis pigmentosa, only a small area of central vision remains, along with slight peripheral vision.
It can be difficult to predict the extent of vision loss or how fast it will progress when you have retinitis pigmentosa. Your eye doctor will likely monitor the health of your retinal cells and administer tests to determine how well you can see.
At some point, you may be advised to drive only during the daytime or on well-lit streets at night. Eventually, you may be unable to see well enough to drive at all.
What causes retinitis pigmentosa?
Rather than being considered a single disease, retinitis pigmentosa is viewed as a group of diseases affecting how light-sensitive cells in the back of the eye function. Not much is known about what causes retinitis pigmentosa, except that the disease is inherited.
The eye condition is associated with at least 32 different genes, which control traits that are passed along in a number of different ways. At times, the genetic trait is dominant and likely to be passed along to a child when a parent has RP. At other times, the trait for retinitis pigmentosa is recessive and may be present for many generations before it appears in a family member.
This means that, even if your mother and father don't have retinitis pigmentosa, you can still have the eye disease when at least one parent carries an altered gene associated with the trait. In fact, about 1% of the population can be considered carriers of genetic traits for retinitis pigmentosa.
Retinitis pigmentosa occurs in about 1 of every 4,000 people in the United States. When the trait is dominant, it is more likely to show up when people are in their 40s. When the trait is recessive, it tends to first appear when people are in their 20s.
Treatments for RP
Currently, there is no cure for retinitis pigmentosa. But several companies are developing retinal implants (sometimes called bionic eyes) and other innovative treatments that are showing promise in providing or preserving some degree of usable sight for people affected by RP.
Retinal Prosthesis System
Implantable retinal prosthesis systems have been developed for patients blinded by retinitis pigmentosa and other degenerative retinal diseases.
These systems typically include a tiny video camera that is built into a special pair of eyeglasses. The camera is connected to a small, wireless device worn by the patient, which coverts the video input to electronic signals that are transmitted wirelessly to the implant in the eye.
The implant uses this information to stimulate remaining healthy cells in the retina, and visual information is thereby transmitted by the optic nerve to the brain, where it is perceived as patterns of light. The patient learns to interpret these patterns so he or she can distinguish the outlines of objects.
At the 2011 annual meeting of the Association for Research in Vision and Ophthalmology (ARVO), researchers presented data from a clinical trial of 30 people blinded by RP or other retinal disease who underwent implantation of one of these early devices at 10 eye surgery centers worldwide over a four-year period.
All patients obtained some visual perceptions from the device, and many showed significant improvements in mobility skills, such as following lines, opening doors and windows and avoiding objects. Two people implanted with the device were able to read short sentences, which exceeded the researchers' expectations, and the gains in vision by many patients were maintained during a follow-up period of at least two years.
Electrical stimulation therapy
For patients with early and intermediate-stage RP, electrical stimulation therapy (EST) of the eye may help preserve vision that otherwise would be lost to the disease.
In a clinical trial of 24 patients with early or mid-stage retinitis pigmentosa that began on 2007, eyes that received small amounts of electrical current delivered to the retina via a tiny electrode showed a significant improvement in field of vision, compared with eyes that did not receive the stimulation.
According to the researchers, the study suggests controlled electrical stimulation of the retina releases growth factors that may delay degeneration of the retina from RP.
Other treatments
Other possible treatments for RP that are being developed include implantable capsules of sustained-release medication that may help preserve or prolong retinal function, vitamin A and other antioxidant nutrient therapies to help reduce retinal damage, and gene therapies designed to transplant normal genes into retinal cells to replace missing or defective ones to potentially halt or reverse retinitis pigmentosa.
Adaptive therapy and low vision devices
Early intervention with occupational therapy may be helpful to manage life changes caused by RP, because it can be easier to adjust to declining vision in earlier stages of vision loss.
Individuals with retinitis pigmentosa also might consider the use of low vision devices that can help magnify and illuminate objects in home and work spaces.